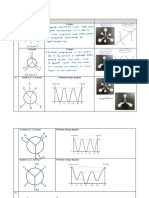
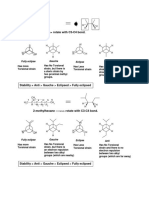
Download as ppt, pdf, or txt
You might also like
- Organic - Class 5 PDFDocument42 pagesOrganic - Class 5 PDFSajan Singh LUCKYNo ratings yet
- Schaum's Easy Outline of Organic Chemistry, Second EditionFrom EverandSchaum's Easy Outline of Organic Chemistry, Second EditionRating: 3.5 out of 5 stars3.5/5 (2)
- Newman Projections Practice Problems-AnswersDocument4 pagesNewman Projections Practice Problems-AnswersNatasha Nadya Hussin100% (1)
- Semester Iv Pharmaceutical Organic Chemistry - Iii (BP401TT) Multiple Choice Questions (Chapter 1 & 2 - Stereochemistry) (Chapter 3 - Heterocyclic Compound - I)Document44 pagesSemester Iv Pharmaceutical Organic Chemistry - Iii (BP401TT) Multiple Choice Questions (Chapter 1 & 2 - Stereochemistry) (Chapter 3 - Heterocyclic Compound - I)Pharma SharmaNo ratings yet
- Hydrocarbons - Halogen Derivatives For JEE Main - JEEced (Study Package For Chemistry) - Dr. O. P. Agarwal PDFDocument326 pagesHydrocarbons - Halogen Derivatives For JEE Main - JEEced (Study Package For Chemistry) - Dr. O. P. Agarwal PDFPaathshala Education IT100% (2)
- Conformational Analysis: Carey & Sundberg: Part A Chapter 3Document53 pagesConformational Analysis: Carey & Sundberg: Part A Chapter 3Dr-Dinesh Kumar100% (1)
- Chem 2Document51 pagesChem 2sunil choudharyNo ratings yet
- L4-L5 Conformational AnalysisDocument60 pagesL4-L5 Conformational Analysisvanwani.mozeelNo ratings yet
- ConformationDocument13 pagesConformationAnand MurugananthamNo ratings yet
- RS Conformational AnalysisL4-L5.Ppt 1Document40 pagesRS Conformational AnalysisL4-L5.Ppt 1Vishal GarimellaNo ratings yet
- 10 Stereo MSC 5Document66 pages10 Stereo MSC 5Gowtham LecturesNo ratings yet
- 9 Stereo MSC 4Document60 pages9 Stereo MSC 4Gowtham LecturesNo ratings yet
- Important Concepts: Conformational Analysis of AlkanesDocument4 pagesImportant Concepts: Conformational Analysis of AlkanesKayla JoezetteNo ratings yet
- Cycloalkanes ConformersDocument15 pagesCycloalkanes ConformersSabila IzzatiNo ratings yet
- Lecture 7 - CyclohexaneDocument9 pagesLecture 7 - Cyclohexanemaishaali2004No ratings yet
- Conform AnalysisDocument3 pagesConform Analysishugo200887No ratings yet
- Chapter 4Document20 pagesChapter 4Simran Saiinii0% (1)
- Stereochemistry (2.1, 2.2)Document12 pagesStereochemistry (2.1, 2.2)VIGHNESH BALKRISHNA LOKARENo ratings yet
- Introduction To Organic Chemistry: C C C CDocument18 pagesIntroduction To Organic Chemistry: C C C CPedro Moreno de SouzaNo ratings yet
- U1 U4有机Document110 pagesU1 U4有机Asick DariusNo ratings yet
- Ch04 Solomon 16-17Document35 pagesCh04 Solomon 16-17HetNo ratings yet
- Confirmation of CyclohexaneDocument3 pagesConfirmation of CyclohexanePARAS GAMERNo ratings yet
- Copy of Manual Amali Sko3023 Sem A222Document5 pagesCopy of Manual Amali Sko3023 Sem A222NURUL ZAKIRAH BINTI BORHANUDINNo ratings yet
- Copy of MANUAL AMALI SKO3023 SEM A222Document5 pagesCopy of MANUAL AMALI SKO3023 SEM A222NURUL ZAKIRAH BINTI BORHANUDINNo ratings yet
- Conformations of Alkanes and CycloalkanesDocument37 pagesConformations of Alkanes and CycloalkanesHelen LugoNo ratings yet
- 8 ConformationalDocument26 pages8 ConformationalAnand MurugananthamNo ratings yet
- Hydrocarbons & Halogen Derivatives PDFDocument326 pagesHydrocarbons & Halogen Derivatives PDFSuraj panditNo ratings yet
- 7.3 Monosubstituted Cyclohexanes. Conformational Analysis: ProblemDocument5 pages7.3 Monosubstituted Cyclohexanes. Conformational Analysis: ProblemenfNo ratings yet
- 1021 Wk5 WS Assessable QuestionsDocument7 pages1021 Wk5 WS Assessable QuestionsGavin DingNo ratings yet
- Anelastic Behavior in Polymers Class NotesDocument18 pagesAnelastic Behavior in Polymers Class NotesShrishty SahuNo ratings yet
- Chapter 2 Lecture Slides PDFDocument108 pagesChapter 2 Lecture Slides PDFjoseph changNo ratings yet
- 1 Semester Organic Chemistry Now in 1 % The Time!Document23 pages1 Semester Organic Chemistry Now in 1 % The Time!Tùng SuNo ratings yet
- DrawingsDocument1 pageDrawingsJAGTAP UTKARSH ASHOKRAONo ratings yet
- Pharmaceutical ChemistryDocument21 pagesPharmaceutical ChemistryMuneneNo ratings yet
- Burrows - Isomerism and Stereochemistry - Cap18Document36 pagesBurrows - Isomerism and Stereochemistry - Cap18FJosue MalaveHNo ratings yet
- Types of Organic IsomerismDocument1 pageTypes of Organic IsomerismAglaete Araújo50% (2)
- Peter Steel S ENCH241 Part1-1Document53 pagesPeter Steel S ENCH241 Part1-1Sam NugentNo ratings yet
- Stereo IsomerismDocument24 pagesStereo IsomerismPardhiv WyattNo ratings yet
- Homework Problems: Structure, Bonding & Hybridization 1. The Molecule Shown Below Is Griseofulvin, An Antifungal CompoundDocument8 pagesHomework Problems: Structure, Bonding & Hybridization 1. The Molecule Shown Below Is Griseofulvin, An Antifungal CompoundPrachi KaushikNo ratings yet
- Alkanes and Cycloalkanes: Organic Chemistry BS Biology - 1 Semester, AY 2020 - 2021Document38 pagesAlkanes and Cycloalkanes: Organic Chemistry BS Biology - 1 Semester, AY 2020 - 2021Rachel AgacoscosNo ratings yet
- Lecture_09_CHE 1502 – General Chemistry 1BDocument42 pagesLecture_09_CHE 1502 – General Chemistry 1BPumza SentseNo ratings yet
- SP1 Stereochemistry PDFDocument66 pagesSP1 Stereochemistry PDFMoulindu KunduNo ratings yet
- Hydrocarbons PDFDocument48 pagesHydrocarbons PDFAniruddha KawadeNo ratings yet
- Conformations of Organic MoleculesDocument21 pagesConformations of Organic MoleculesAkash KolliNo ratings yet
- Stereo ChemistryDocument15 pagesStereo ChemistryhamsiniyvreddyNo ratings yet
- Dragojlovic2015 Article ConformationalAnalysisOfCycloaDocument30 pagesDragojlovic2015 Article ConformationalAnalysisOfCycloaNimra MalikNo ratings yet
- 2Document41 pages2Shravani KhotNo ratings yet
- Isomerism: Definition-Structural Isomers: Same Molecular Formula Different Structures (Or Structural Formulae)Document13 pagesIsomerism: Definition-Structural Isomers: Same Molecular Formula Different Structures (Or Structural Formulae)Trần Duy Tân100% (1)
- Topic 4.6 Aromatic ChemistryDocument15 pagesTopic 4.6 Aromatic ChemistrySantanu PachhalNo ratings yet
- Topic 18 NotesDocument12 pagesTopic 18 NotessherkhanNo ratings yet
- Lec 2 - MCDocument22 pagesLec 2 - MCDivyam JainNo ratings yet
- Confirmations of CyclohexaneDocument15 pagesConfirmations of CyclohexaneSuliman WardakNo ratings yet
- Conformation: Just When You Thought It Was Safe To Look at Molecules Again... Came Shape!Document106 pagesConformation: Just When You Thought It Was Safe To Look at Molecules Again... Came Shape!YoAmoNYC100% (1)
- Butane - Rotate With C3-C4 Bond.: C H C H C H CH H H H HDocument11 pagesButane - Rotate With C3-C4 Bond.: C H C H C H CH H H H HMadison FullerNo ratings yet
- Hydrocarbons Final PDFDocument74 pagesHydrocarbons Final PDFAnurag SahuNo ratings yet
- Final Exam, Key M2011ADocument27 pagesFinal Exam, Key M2011AThanh VyNo ratings yet
- CH CH CHCH CH H CH CH CH CH CH CH H CH: Byvineet Khatri SirDocument13 pagesCH CH CHCH CH H CH CH CH CH CH CH H CH: Byvineet Khatri Sirsarvesh goyalNo ratings yet
- Electron Pushing PDFDocument3 pagesElectron Pushing PDFGiacomo Dal PraNo ratings yet
- Intermolecular Forces Group Exercise - CHEM 1140Document3 pagesIntermolecular Forces Group Exercise - CHEM 1140Redzwan KadirNo ratings yet
- 12 - Alkanes Lecture NotesDocument15 pages12 - Alkanes Lecture Notesقاتل مستأجرNo ratings yet
- Homework 3 - STEREOCHEMISTRY AT TETRAHEDRAL CENTERSDocument3 pagesHomework 3 - STEREOCHEMISTRY AT TETRAHEDRAL CENTERSThảo UyênNo ratings yet
- 15 Types of IsomersDocument4 pages15 Types of IsomersGhady AbdellatifNo ratings yet
- Cyclo Alkanes CompltDocument8 pagesCyclo Alkanes CompltKaneez AsmaNo ratings yet
- Stereo NotesDocument37 pagesStereo NotesSankar AdhikariNo ratings yet
- Chemistry: Isomerism Theory & QuestionsDocument124 pagesChemistry: Isomerism Theory & Questionsgaurav nigamNo ratings yet
- CHM01 CO3 LESSON2 Molecular-ShapesDocument14 pagesCHM01 CO3 LESSON2 Molecular-ShapesErica MamauagNo ratings yet
- CML-101-Tutorial 1 - Anwser KeyDocument10 pagesCML-101-Tutorial 1 - Anwser KeySarthakNo ratings yet
- 化學奧林匹亞冬令營 有機3 2018Document119 pages化學奧林匹亞冬令營 有機3 2018楊泰萱No ratings yet
- Pharmaceutical Organic Chemistry-Iii: Instructions To CandidatesDocument2 pagesPharmaceutical Organic Chemistry-Iii: Instructions To CandidatesNitish PathaniaNo ratings yet
- Notes 20 (3) Stereoisomers HL ONLYDocument8 pagesNotes 20 (3) Stereoisomers HL ONLYmickey mouseNo ratings yet
- Stereochemical Aspects of Organic SynthesisDocument9 pagesStereochemical Aspects of Organic SynthesisNoor NNo ratings yet
- Single Choice Question 1.: H CH CH H H CH CH CH H CH CH CH H CHDocument7 pagesSingle Choice Question 1.: H CH CH H H CH CH CH H CH CH CH H CHSumit RajNo ratings yet
- Asymmetric Synthesis: Presented By: Faizan Ahmed M.SC (F) Presented To: Dr. Imran Ali HashmiDocument39 pagesAsymmetric Synthesis: Presented By: Faizan Ahmed M.SC (F) Presented To: Dr. Imran Ali HashmiAndré Luiz Meleiro PortoNo ratings yet
- IsomerismDocument16 pagesIsomerismcvjaykumarreddy100% (2)
- Prep 101 Booklet (2013) Part 2Document24 pagesPrep 101 Booklet (2013) Part 2Alexandre SaymanNo ratings yet
- Experiment 1 SkoDocument4 pagesExperiment 1 SkoLau Lee Ling100% (2)
- L1-L3 StereochemistryDocument64 pagesL1-L3 Stereochemistryvanwani.mozeelNo ratings yet
- Chiral Centre and CIP Nomenclature Stereochemistry by Rajeev Gupta Organic ChemistryDocument13 pagesChiral Centre and CIP Nomenclature Stereochemistry by Rajeev Gupta Organic ChemistryDr. Rajeev GuptaNo ratings yet
- Nomenclature, Bonding, and Isomers FlashcardsDocument106 pagesNomenclature, Bonding, and Isomers FlashcardsLejNo ratings yet
- ICT BHB Sem 2 2Document59 pagesICT BHB Sem 2 2Ayushmaan TripathiNo ratings yet
- Monosaccharides:, Dr. Shweta HardiaDocument28 pagesMonosaccharides:, Dr. Shweta HardiaArzoo RathoreNo ratings yet
- CML 100 Questions PDFDocument7 pagesCML 100 Questions PDFDivyansh GuptaNo ratings yet
- Nitrogen Inversion: What Is It?Document4 pagesNitrogen Inversion: What Is It?Soham ChakrabortyNo ratings yet
- Stereo ChemistryDocument34 pagesStereo ChemistrylNo ratings yet
- Chapter 2-StereochemistryDocument26 pagesChapter 2-Stereochemistrychalagirma95No ratings yet
- Cyclic Alkanes & AlkenesDocument18 pagesCyclic Alkanes & AlkenesbgbscgvNo ratings yet
- Chapter 03Document40 pagesChapter 03AC BañaresNo ratings yet
- Chirality DDDocument130 pagesChirality DDRahul Doc0% (1)