
Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia
You might also like
- LymphomaDocument78 pagesLymphomaastriwahyuni100% (4)
- Katalog - Polyester Distribution Boxes BARTECDocument4 pagesKatalog - Polyester Distribution Boxes BARTECPedjaNo ratings yet
- Leucemia Limfatica CronicaDocument52 pagesLeucemia Limfatica CronicaAnghel BogdanNo ratings yet
- Klasifikasi LimfomaDocument5 pagesKlasifikasi LimfomaBadlina Fitrianisa YulianingrumNo ratings yet
- Curs5 Hematologie AnvDocument59 pagesCurs5 Hematologie AnvRaluca PăunaNo ratings yet
- Lymphoma Myelofibrosis LectDocument99 pagesLymphoma Myelofibrosis LectShiv SinghNo ratings yet
- Hem/Onc USML Organization ChartDocument1 pageHem/Onc USML Organization ChartJJNo ratings yet
- Boala HodgkinDocument44 pagesBoala HodgkinCristina BuzatuNo ratings yet
- 2 NHLDocument13 pages2 NHLwie_wie_wieNo ratings yet
- NHL FK UnimalDocument46 pagesNHL FK UnimalicaNo ratings yet
- Non-Hodgkins LymphomaDocument22 pagesNon-Hodgkins LymphomaNyoman TapayanaNo ratings yet
- C6.1clasificare LimfoameDocument10 pagesC6.1clasificare LimfoameRădulescu AndreeaNo ratings yet
- Bolile Mieloproliferative CroniceDocument57 pagesBolile Mieloproliferative Cronicetrifex100% (2)
- LMC CursDocument31 pagesLMC CursEma ȚurcașNo ratings yet
- Lymph Node CytologyDocument39 pagesLymph Node Cytologykamranghani641No ratings yet
- Non Hodgkin's LymphomaDocument31 pagesNon Hodgkin's LymphomaJeo Thomas100% (1)
- Lymphoid NeoplasmsDocument52 pagesLymphoid NeoplasmsAmalia Riska GNo ratings yet
- Non-Hodgkin's Lymphoma: Luis Fayad, MDDocument13 pagesNon-Hodgkin's Lymphoma: Luis Fayad, MDafdhonaNo ratings yet
- 2 NHLDocument13 pages2 NHLJefry SNo ratings yet
- Prof. Dr. Gamal Abdul Hamid University of AdenDocument95 pagesProf. Dr. Gamal Abdul Hamid University of AdenAarpit PatwaNo ratings yet
- Acute Leukaemias: DR Nilukshi Perera Consultant HaematologistDocument60 pagesAcute Leukaemias: DR Nilukshi Perera Consultant HaematologistThaveeshaLindsayWhite100% (2)
- Malignant Lymphomas: DR Nilukshi PereraDocument48 pagesMalignant Lymphomas: DR Nilukshi PereraThaveeshaLindsayWhiteNo ratings yet
- Leukemia: Dr. Suhaemi, SPPD, FinasimDocument96 pagesLeukemia: Dr. Suhaemi, SPPD, Finasimwie_wie_wieNo ratings yet
- Chapter 19 20 LymphomaDocument30 pagesChapter 19 20 Lymphomaehabeideh11No ratings yet
- Acute Lymphoblastic LeukemiaDocument27 pagesAcute Lymphoblastic LeukemiaMahalakshmi PalanisamiNo ratings yet
- Acute LeukaemiaDocument39 pagesAcute LeukaemiaAbby LiewNo ratings yet
- CFU: Colony Forming Unit: Ploripotent Stem CellDocument1 pageCFU: Colony Forming Unit: Ploripotent Stem CellFlorentina Lisa PratamaNo ratings yet
- Haematologic MalignanciesDocument18 pagesHaematologic MalignanciesMukesh SahooNo ratings yet
- ASH Hematology Review Series - Indolent LymphomasDocument77 pagesASH Hematology Review Series - Indolent LymphomasИван НегарэNo ratings yet
- Acute Leukemia: David Lee, MD, FRCPCDocument31 pagesAcute Leukemia: David Lee, MD, FRCPCfranzzjosefNo ratings yet
- LeukemiaDocument26 pagesLeukemiaochamocha100% (1)
- 4b TUMOR JAR RetikuloendotelialAAADocument98 pages4b TUMOR JAR RetikuloendotelialAAARyo RyozNo ratings yet
- Acute Leukaemia-Update: DR Niranjan N. RathodDocument89 pagesAcute Leukaemia-Update: DR Niranjan N. RathodratanNo ratings yet
- Leukemia: Dr. Isbandiyah SPPD Bag. Ilmu Penyakit Dalam Umm MalangDocument26 pagesLeukemia: Dr. Isbandiyah SPPD Bag. Ilmu Penyakit Dalam Umm MalangSemesta0% (1)
- Vasile Musteata, MD, PHD, MPH, Associate Professor Discipline of Hematology, State University of Medicine and Pharmacy "N. Testemitanu"Document49 pagesVasile Musteata, MD, PHD, MPH, Associate Professor Discipline of Hematology, State University of Medicine and Pharmacy "N. Testemitanu"AlbuNo ratings yet
- LeukemiaDocument26 pagesLeukemiameutia salsabilaNo ratings yet
- Malignant HematologyDocument1 pageMalignant HematologyaustinlwongNo ratings yet
- Chart - WBC DisordersDocument1 pageChart - WBC DisordersSamuel RothschildNo ratings yet
- Elearning Lymphatic Organs 2 2023newDocument28 pagesElearning Lymphatic Organs 2 2023newpiano357sidNo ratings yet
- Acute Myeloproliferative Acute Lymphoproliferative Chronic Myeloproliferative Chronic Lymphoproliferative Plasma Cell NeoplasmDocument1 pageAcute Myeloproliferative Acute Lymphoproliferative Chronic Myeloproliferative Chronic Lymphoproliferative Plasma Cell NeoplasmAudreySlitNo ratings yet
- WBC Neoplasms Review - PathologyDocument6 pagesWBC Neoplasms Review - Pathologylas100% (6)
- Hematopatologie 2016Document45 pagesHematopatologie 2016CristiNo ratings yet
- Blockxiv Neoplasms Lymphoid 2006Document54 pagesBlockxiv Neoplasms Lymphoid 2006Ryo RyozNo ratings yet
- Leucemia Limfatica CronicaDocument18 pagesLeucemia Limfatica CronicaUngureanu Andrei100% (1)
- Malignant Disorders of Leukocytes: Supachai A. Basit, RMT, PHDDocument109 pagesMalignant Disorders of Leukocytes: Supachai A. Basit, RMT, PHDChatie PipitNo ratings yet
- Leukemia AcuteDocument7 pagesLeukemia AcutefallstarrNo ratings yet
- 20 Lymphomas and LeukemiastextsDocument40 pages20 Lymphomas and LeukemiastextsArief SeptianurNo ratings yet
- Acute Leukemias: Atu Level 300 Practical SectionDocument35 pagesAcute Leukemias: Atu Level 300 Practical SectionAbdul Raouf KhalidNo ratings yet
- Leukemias and Lymphomas Flow Chart ModifiedDocument5 pagesLeukemias and Lymphomas Flow Chart Modifiedlovelyc95No ratings yet
- Hematopathlogy 2Document87 pagesHematopathlogy 2evansmando12No ratings yet
- LeukemiaDocument40 pagesLeukemiaYeni Jesika SilitongaNo ratings yet
- LNs HNDocument190 pagesLNs HNNinna Isabel VictorioNo ratings yet
- Notes, 1/e: Plasma Cell TumorsDocument9 pagesNotes, 1/e: Plasma Cell TumorsvkNo ratings yet
- 18 Characteristics of Leukemias Lymphomas and MyelomasDocument9 pages18 Characteristics of Leukemias Lymphomas and MyelomasDaphne HernaezNo ratings yet
- Cat HyperlymphocytoseDocument40 pagesCat HyperlymphocytoseDésiré Pengdéwendé OUEDRAOGONo ratings yet
- Hemopoietic Disorders IIDocument23 pagesHemopoietic Disorders IIAnn patricia ViolaNo ratings yet
- WBC DisordersDocument114 pagesWBC DisordersNdor BariboloNo ratings yet
- Rope PDFDocument19 pagesRope PDFBenjamin van DierenNo ratings yet
- The First Vertebrates, Jawless Fishes, The Agnathans: 2.1 OstracodermsDocument22 pagesThe First Vertebrates, Jawless Fishes, The Agnathans: 2.1 OstracodermsAlejandro Tepoz TelloNo ratings yet
- Urushi ArtDocument24 pagesUrushi ArtGuadalupeCaravajalNo ratings yet
- OpenFOAM编程指南Document100 pagesOpenFOAM编程指南Feishi XuNo ratings yet
- Ex5500 PDFDocument7 pagesEx5500 PDFRoberto Chang PalmaNo ratings yet
- Protocols For Public-Key CryptosystemsDocument13 pagesProtocols For Public-Key CryptosystemsIvo LemosNo ratings yet
- Fallas VenezuelaDocument20 pagesFallas VenezuelaDaniel Quintana GaviriaNo ratings yet
- Stream User-ManualDocument36 pagesStream User-ManualSreekanth NakkaNo ratings yet
- Power Electronics: Thyristor Controlled Power For Electric MotorsDocument10 pagesPower Electronics: Thyristor Controlled Power For Electric Motorsshahab moinNo ratings yet
- Reading Test 1: Questions 1-5 Refer To The Following ArticleDocument14 pagesReading Test 1: Questions 1-5 Refer To The Following ArticletrucNo ratings yet
- 2018 Weekly CalendarDocument3 pages2018 Weekly CalendarFabian FebianoNo ratings yet
- Native Son Essay TopicsDocument7 pagesNative Son Essay TopicsafabioemwNo ratings yet
- Written RequestDocument2 pagesWritten Requestcarvazro100% (1)
- Gen Studs and Engg AptiDocument1 pageGen Studs and Engg AptiasishNo ratings yet
- Jambajuicelv-Application-0618 1Document2 pagesJambajuicelv-Application-0618 1api-526082107No ratings yet
- Thrift Banks ActDocument25 pagesThrift Banks ActMadelle Pineda100% (1)
- Turbine-Less Ducted Fan Jet Engine: Subsonic PropulsionDocument25 pagesTurbine-Less Ducted Fan Jet Engine: Subsonic PropulsionزهديابوانسNo ratings yet
- Английская грамматика в тестах - РомановаDocument152 pagesАнглийская грамматика в тестах - РомановаLi FeNo ratings yet
- Task 6 - Leave Type Safety ValveDocument3 pagesTask 6 - Leave Type Safety ValveTeguh RaharjoNo ratings yet
- Parasnis - 1951 - Study Rock MidlandsDocument20 pagesParasnis - 1951 - Study Rock MidlandsIsaac KandaNo ratings yet
- FACTORY IO-Sorting of Boxes (1) / PLC - 1 (CPU 1212C AC/DC/Rly) / Pro Gram BlocksDocument3 pagesFACTORY IO-Sorting of Boxes (1) / PLC - 1 (CPU 1212C AC/DC/Rly) / Pro Gram BlocksHasaan HussainNo ratings yet
- Transistor 2n3904 DatasheetDocument2 pagesTransistor 2n3904 DatasheetAlex ZXNo ratings yet
- Title of Project:-Military Hospital Report Management SystemDocument4 pagesTitle of Project:-Military Hospital Report Management SystemAkbar AliNo ratings yet
- Ozone Therapy Is Safest Known TherapyDocument35 pagesOzone Therapy Is Safest Known Therapyherdin2008100% (1)
- Activity Sheets Signal WordsDocument15 pagesActivity Sheets Signal WordsGrace Ann EscabarteNo ratings yet
- Laboratorium Pengujian Teknik Sipil Universitas Bandar LampungDocument1 pageLaboratorium Pengujian Teknik Sipil Universitas Bandar LampungPanji OctaWirawanNo ratings yet
- 21CC 4 U6Document10 pages21CC 4 U6Adrian Spanu100% (1)
- Norton TheoremDocument18 pagesNorton TheoremZohaib NasirNo ratings yet
- Kargil Facts - Indian Soldiers Were RAPED, Then CASTRATED by Pakistani ArmyDocument6 pagesKargil Facts - Indian Soldiers Were RAPED, Then CASTRATED by Pakistani Armyhindu.nation100% (1)






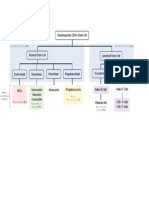



















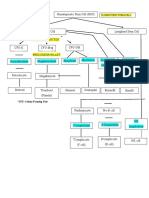































































Download as pptx, pdf, or txt
You might also like
- LymphomaDocument78 pagesLymphomaastriwahyuni100% (4)
- Katalog - Polyester Distribution Boxes BARTECDocument4 pagesKatalog - Polyester Distribution Boxes BARTECPedjaNo ratings yet
- Leucemia Limfatica CronicaDocument52 pagesLeucemia Limfatica CronicaAnghel BogdanNo ratings yet
- Klasifikasi LimfomaDocument5 pagesKlasifikasi LimfomaBadlina Fitrianisa YulianingrumNo ratings yet
- Curs5 Hematologie AnvDocument59 pagesCurs5 Hematologie AnvRaluca PăunaNo ratings yet
- Lymphoma Myelofibrosis LectDocument99 pagesLymphoma Myelofibrosis LectShiv SinghNo ratings yet
- Hem/Onc USML Organization ChartDocument1 pageHem/Onc USML Organization ChartJJNo ratings yet
- Boala HodgkinDocument44 pagesBoala HodgkinCristina BuzatuNo ratings yet
- 2 NHLDocument13 pages2 NHLwie_wie_wieNo ratings yet
- NHL FK UnimalDocument46 pagesNHL FK UnimalicaNo ratings yet
- Non-Hodgkins LymphomaDocument22 pagesNon-Hodgkins LymphomaNyoman TapayanaNo ratings yet
- C6.1clasificare LimfoameDocument10 pagesC6.1clasificare LimfoameRădulescu AndreeaNo ratings yet
- Bolile Mieloproliferative CroniceDocument57 pagesBolile Mieloproliferative Cronicetrifex100% (2)
- LMC CursDocument31 pagesLMC CursEma ȚurcașNo ratings yet
- Lymph Node CytologyDocument39 pagesLymph Node Cytologykamranghani641No ratings yet
- Non Hodgkin's LymphomaDocument31 pagesNon Hodgkin's LymphomaJeo Thomas100% (1)
- Lymphoid NeoplasmsDocument52 pagesLymphoid NeoplasmsAmalia Riska GNo ratings yet
- Non-Hodgkin's Lymphoma: Luis Fayad, MDDocument13 pagesNon-Hodgkin's Lymphoma: Luis Fayad, MDafdhonaNo ratings yet
- 2 NHLDocument13 pages2 NHLJefry SNo ratings yet
- Prof. Dr. Gamal Abdul Hamid University of AdenDocument95 pagesProf. Dr. Gamal Abdul Hamid University of AdenAarpit PatwaNo ratings yet
- Acute Leukaemias: DR Nilukshi Perera Consultant HaematologistDocument60 pagesAcute Leukaemias: DR Nilukshi Perera Consultant HaematologistThaveeshaLindsayWhite100% (2)
- Malignant Lymphomas: DR Nilukshi PereraDocument48 pagesMalignant Lymphomas: DR Nilukshi PereraThaveeshaLindsayWhiteNo ratings yet
- Leukemia: Dr. Suhaemi, SPPD, FinasimDocument96 pagesLeukemia: Dr. Suhaemi, SPPD, Finasimwie_wie_wieNo ratings yet
- Chapter 19 20 LymphomaDocument30 pagesChapter 19 20 Lymphomaehabeideh11No ratings yet
- Acute Lymphoblastic LeukemiaDocument27 pagesAcute Lymphoblastic LeukemiaMahalakshmi PalanisamiNo ratings yet
- Acute LeukaemiaDocument39 pagesAcute LeukaemiaAbby LiewNo ratings yet
- CFU: Colony Forming Unit: Ploripotent Stem CellDocument1 pageCFU: Colony Forming Unit: Ploripotent Stem CellFlorentina Lisa PratamaNo ratings yet
- Haematologic MalignanciesDocument18 pagesHaematologic MalignanciesMukesh SahooNo ratings yet
- ASH Hematology Review Series - Indolent LymphomasDocument77 pagesASH Hematology Review Series - Indolent LymphomasИван НегарэNo ratings yet
- Acute Leukemia: David Lee, MD, FRCPCDocument31 pagesAcute Leukemia: David Lee, MD, FRCPCfranzzjosefNo ratings yet
- LeukemiaDocument26 pagesLeukemiaochamocha100% (1)
- 4b TUMOR JAR RetikuloendotelialAAADocument98 pages4b TUMOR JAR RetikuloendotelialAAARyo RyozNo ratings yet
- Acute Leukaemia-Update: DR Niranjan N. RathodDocument89 pagesAcute Leukaemia-Update: DR Niranjan N. RathodratanNo ratings yet
- Leukemia: Dr. Isbandiyah SPPD Bag. Ilmu Penyakit Dalam Umm MalangDocument26 pagesLeukemia: Dr. Isbandiyah SPPD Bag. Ilmu Penyakit Dalam Umm MalangSemesta0% (1)
- Vasile Musteata, MD, PHD, MPH, Associate Professor Discipline of Hematology, State University of Medicine and Pharmacy "N. Testemitanu"Document49 pagesVasile Musteata, MD, PHD, MPH, Associate Professor Discipline of Hematology, State University of Medicine and Pharmacy "N. Testemitanu"AlbuNo ratings yet
- LeukemiaDocument26 pagesLeukemiameutia salsabilaNo ratings yet
- Malignant HematologyDocument1 pageMalignant HematologyaustinlwongNo ratings yet
- Chart - WBC DisordersDocument1 pageChart - WBC DisordersSamuel RothschildNo ratings yet
- Elearning Lymphatic Organs 2 2023newDocument28 pagesElearning Lymphatic Organs 2 2023newpiano357sidNo ratings yet
- Acute Myeloproliferative Acute Lymphoproliferative Chronic Myeloproliferative Chronic Lymphoproliferative Plasma Cell NeoplasmDocument1 pageAcute Myeloproliferative Acute Lymphoproliferative Chronic Myeloproliferative Chronic Lymphoproliferative Plasma Cell NeoplasmAudreySlitNo ratings yet
- WBC Neoplasms Review - PathologyDocument6 pagesWBC Neoplasms Review - Pathologylas100% (6)
- Hematopatologie 2016Document45 pagesHematopatologie 2016CristiNo ratings yet
- Blockxiv Neoplasms Lymphoid 2006Document54 pagesBlockxiv Neoplasms Lymphoid 2006Ryo RyozNo ratings yet
- Leucemia Limfatica CronicaDocument18 pagesLeucemia Limfatica CronicaUngureanu Andrei100% (1)
- Malignant Disorders of Leukocytes: Supachai A. Basit, RMT, PHDDocument109 pagesMalignant Disorders of Leukocytes: Supachai A. Basit, RMT, PHDChatie PipitNo ratings yet
- Leukemia AcuteDocument7 pagesLeukemia AcutefallstarrNo ratings yet
- 20 Lymphomas and LeukemiastextsDocument40 pages20 Lymphomas and LeukemiastextsArief SeptianurNo ratings yet
- Acute Leukemias: Atu Level 300 Practical SectionDocument35 pagesAcute Leukemias: Atu Level 300 Practical SectionAbdul Raouf KhalidNo ratings yet
- Leukemias and Lymphomas Flow Chart ModifiedDocument5 pagesLeukemias and Lymphomas Flow Chart Modifiedlovelyc95No ratings yet
- Hematopathlogy 2Document87 pagesHematopathlogy 2evansmando12No ratings yet
- LeukemiaDocument40 pagesLeukemiaYeni Jesika SilitongaNo ratings yet
- LNs HNDocument190 pagesLNs HNNinna Isabel VictorioNo ratings yet
- Notes, 1/e: Plasma Cell TumorsDocument9 pagesNotes, 1/e: Plasma Cell TumorsvkNo ratings yet
- 18 Characteristics of Leukemias Lymphomas and MyelomasDocument9 pages18 Characteristics of Leukemias Lymphomas and MyelomasDaphne HernaezNo ratings yet
- Cat HyperlymphocytoseDocument40 pagesCat HyperlymphocytoseDésiré Pengdéwendé OUEDRAOGONo ratings yet
- Hemopoietic Disorders IIDocument23 pagesHemopoietic Disorders IIAnn patricia ViolaNo ratings yet
- WBC DisordersDocument114 pagesWBC DisordersNdor BariboloNo ratings yet
- Rope PDFDocument19 pagesRope PDFBenjamin van DierenNo ratings yet
- The First Vertebrates, Jawless Fishes, The Agnathans: 2.1 OstracodermsDocument22 pagesThe First Vertebrates, Jawless Fishes, The Agnathans: 2.1 OstracodermsAlejandro Tepoz TelloNo ratings yet
- Urushi ArtDocument24 pagesUrushi ArtGuadalupeCaravajalNo ratings yet
- OpenFOAM编程指南Document100 pagesOpenFOAM编程指南Feishi XuNo ratings yet
- Ex5500 PDFDocument7 pagesEx5500 PDFRoberto Chang PalmaNo ratings yet
- Protocols For Public-Key CryptosystemsDocument13 pagesProtocols For Public-Key CryptosystemsIvo LemosNo ratings yet
- Fallas VenezuelaDocument20 pagesFallas VenezuelaDaniel Quintana GaviriaNo ratings yet
- Stream User-ManualDocument36 pagesStream User-ManualSreekanth NakkaNo ratings yet
- Power Electronics: Thyristor Controlled Power For Electric MotorsDocument10 pagesPower Electronics: Thyristor Controlled Power For Electric Motorsshahab moinNo ratings yet
- Reading Test 1: Questions 1-5 Refer To The Following ArticleDocument14 pagesReading Test 1: Questions 1-5 Refer To The Following ArticletrucNo ratings yet
- 2018 Weekly CalendarDocument3 pages2018 Weekly CalendarFabian FebianoNo ratings yet
- Native Son Essay TopicsDocument7 pagesNative Son Essay TopicsafabioemwNo ratings yet
- Written RequestDocument2 pagesWritten Requestcarvazro100% (1)
- Gen Studs and Engg AptiDocument1 pageGen Studs and Engg AptiasishNo ratings yet
- Jambajuicelv-Application-0618 1Document2 pagesJambajuicelv-Application-0618 1api-526082107No ratings yet
- Thrift Banks ActDocument25 pagesThrift Banks ActMadelle Pineda100% (1)
- Turbine-Less Ducted Fan Jet Engine: Subsonic PropulsionDocument25 pagesTurbine-Less Ducted Fan Jet Engine: Subsonic PropulsionزهديابوانسNo ratings yet
- Английская грамматика в тестах - РомановаDocument152 pagesАнглийская грамматика в тестах - РомановаLi FeNo ratings yet
- Task 6 - Leave Type Safety ValveDocument3 pagesTask 6 - Leave Type Safety ValveTeguh RaharjoNo ratings yet
- Parasnis - 1951 - Study Rock MidlandsDocument20 pagesParasnis - 1951 - Study Rock MidlandsIsaac KandaNo ratings yet
- FACTORY IO-Sorting of Boxes (1) / PLC - 1 (CPU 1212C AC/DC/Rly) / Pro Gram BlocksDocument3 pagesFACTORY IO-Sorting of Boxes (1) / PLC - 1 (CPU 1212C AC/DC/Rly) / Pro Gram BlocksHasaan HussainNo ratings yet
- Transistor 2n3904 DatasheetDocument2 pagesTransistor 2n3904 DatasheetAlex ZXNo ratings yet
- Title of Project:-Military Hospital Report Management SystemDocument4 pagesTitle of Project:-Military Hospital Report Management SystemAkbar AliNo ratings yet
- Ozone Therapy Is Safest Known TherapyDocument35 pagesOzone Therapy Is Safest Known Therapyherdin2008100% (1)
- Activity Sheets Signal WordsDocument15 pagesActivity Sheets Signal WordsGrace Ann EscabarteNo ratings yet
- Laboratorium Pengujian Teknik Sipil Universitas Bandar LampungDocument1 pageLaboratorium Pengujian Teknik Sipil Universitas Bandar LampungPanji OctaWirawanNo ratings yet
- 21CC 4 U6Document10 pages21CC 4 U6Adrian Spanu100% (1)
- Norton TheoremDocument18 pagesNorton TheoremZohaib NasirNo ratings yet
- Kargil Facts - Indian Soldiers Were RAPED, Then CASTRATED by Pakistani ArmyDocument6 pagesKargil Facts - Indian Soldiers Were RAPED, Then CASTRATED by Pakistani Armyhindu.nation100% (1)